Определение подобия между двумя последовательностями является общей задачей в вычислительной биологии. Начиная с последовательности нуклеотида для человеческого гена этот пример использует алгоритмы выравнивания, чтобы определить местоположение и проверить соответствующий ген в организме модели.
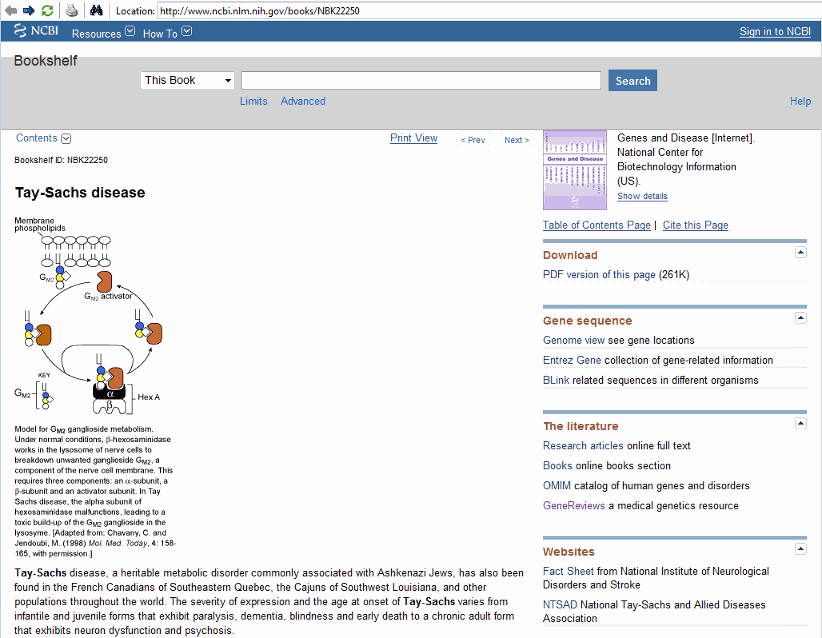
В этом примере вы интересуетесь учащейся болезнью Тея-Сакса. Болезнь Тея-Сакса является автосомальной удаляющейся болезнью, вызванной отсутствием беты-hexosaminidase A фермента (Шестнадцатеричное число А). Этот фермент ответственен за отказ ганглиозидов (GM2) в клетках головного мозга и нервных клетках.
Во-первых, исследовательская информация о болезни Тея-Сакса и ферменте, который сопоставлен с этой болезнью, затем найдите последовательность нуклеотида для человеческого гена, который коды для фермента, и наконец находят, что соответствующий ген в другом организме использует в качестве модели для исследования.
Используйте Браузер документации MATLAB®, чтобы исследовать сеть. В Командном Окне MATLAB введите
web('http://www.ncbi.nlm.nih.gov/books/NBK22250/')
Браузер документации MATLAB открывается страницей болезни Тея-Сакса в разделе Genes и Diseases веб-сайта NCBI. Этот раздел обеспечивает всестороннее введение в медицинскую генетику. В частности, эта страница содержит введение и графическое представление Шестнадцатеричного числа фермента А и его роли в метаболизме липида ганглиозид GM2.
После завершения вашего исследования вы завершили следующее:
Ген коды HEXA для альфа-подблока фермента димера hexosaminidase (Шестнадцатеричное число А), в то время как ген коды HEXB для бета подблока фермента. Третий ген, GM2A, коды для белка активатора GM2. Однако это - мутация в гене HEXA, который вызывает болезнь Тея-Сакса.
Следующая процедура иллюстрирует, как найти последовательность нуклеотида для человеческого гена в общедоступной базе данных и считать информации последовательности в среду MATLAB. Много общедоступных баз данных для последовательностей нуклеотида (например, GenBank®, EMBL-EBI) доступны с сети. Окно Команды MATLAB с Браузером документации MATLAB обеспечивает интегрированную среду для того, чтобы искать в Интернете и принести информацию о последовательности в среду MATLAB.
После того, как вы определите местоположение последовательности, необходимо переместить данные о последовательности в рабочее пространство MATLAB.
Откройте Браузер документации MATLAB для веб-сайта NCBI. В Висячей строке Команды MATLAB ввести
web('http://www.ncbi.nlm.nih.gov/')
Окно Браузера документации MATLAB открывается домашней страницей NCBI.
Ищите ген, вы интересуетесь изучением. Например, из Поискового списка, выберите Nucleotide, и в для поля вводят Tay-Sachs.
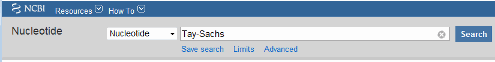
Поиск возвращает записи для генов, которые кодируют альфу и бета подблоки фермента hexosaminidase (Шестнадцатеричное число А) и ген, который кодирует фермент активатора. Ссылка NCBI для человеческого гена HEXA имеет инвентарный номер NM_000520.
Получите данные о последовательности в среду MATLAB. Например, чтобы получить информацию последовательности для человеческого гена HEXA, ввести
humanHEXA = getgenbank('NM_000520')
Пробелы в инвентарных номерах GenBank используют символ подчеркивания. Ввод 'NM 00520' возвращает неправильную запись.
Человеческий ген загружается в рабочее пространство MATLAB как структура.
humanHEXA =
LocusName: 'NM_000520'
LocusSequenceLength: '2255'
LocusNumberofStrands: ''
LocusTopology: 'linear'
LocusMoleculeType: 'mRNA'
LocusGenBankDivision: 'PRI'
LocusModificationDate: '13-AUG-2006'
Definition: 'Homo sapiens hexosaminidase A (alpha polypeptide) (HEXA), mRNA.'
Accession: 'NM_000520'
Version: 'NM_000520.2'
GI: '13128865'
Project: []
Keywords: []
Segment: []
Source: 'Homo sapiens (human)'
SourceOrganism: [4x65 char]
Reference: {1x58 cell}
Comment: [15x67 char]
Features: [74x74 char]
CDS: [1x1 struct]
Sequence: [1x2255 char]
SearchURL: [1x108 char]
RetrieveURL: [1x97 char]
Следующая процедура иллюстрирует, как найти последовательность нуклеотида для гена мыши связанной с человеческим геном, и считайте информации последовательности в среду MATLAB. Последовательность и функция многих генов сохраняются во время эволюции разновидностей через гомологичные гены. Гомологичные гены являются генами, которые имеют общего предка и подобные последовательности. Одна цель поиска общедоступной базы данных состоит в том, чтобы найти подобные гены. Если вы можете определить местоположение последовательности в базе данных, которая похожа на ваш неизвестный ген или белок, вероятно, что функция и характеристики известных и неизвестных генов являются тем же самым.
После нахождения последовательности нуклеотида для человеческого гена можно сделать поиск BLAST или поиск в геноме другого организма для соответствующего гена. Эта процедура использует геном мыши в качестве примера.
Откройте Браузер документации MATLAB для веб-сайта NCBI. В Командном Окне MATLAB введите
web('http://www.ncbi.nlm.nih.gov')
Ищите базу данных нуклеотида ген или белок, вы интересуетесь изучением. Например, из Поискового списка, выберите Nucleotide, и в для поля вводят hexosaminidase A.
Поиск возвращает записи для мыши и геномов человека. Ссылка NCBI для гена мыши HEXA имеет инвентарный номер AK080777.

Получите информацию последовательности для гена мыши в среду MATLAB. Ввод
mouseHEXA = getgenbank('AK080777')
Последовательность генов мыши загружается в рабочее пространство MATLAB как структура.
mouseHEXA =
LocusName: 'AK080777'
LocusSequenceLength: '1839'
LocusNumberofStrands: ''
LocusTopology: 'linear'
LocusMoleculeType: 'mRNA'
LocusGenBankDivision: 'HTC'
LocusModificationDate: '02-SEP-2005'
Definition: [1x150 char]
Accession: 'AK080777'
Version: 'AK080777.1'
GI: '26348756'
Project: []
Keywords: 'HTC; CAP trapper.'
Segment: []
Source: 'Mus musculus (house mouse)'
SourceOrganism: [4x65 char]
Reference: {1x8 cell}
Comment: [8x66 char]
Features: [33x74 char]
CDS: [1x1 struct]
Sequence: [1x1839 char]
SearchURL: [1x107 char]
RetrieveURL: [1x97 char]
Следующая процедура иллюстрирует, как преобразовать последовательность от нуклеотидов до аминокислот и идентифицировать открытые рамки считывания. Последовательность нуклеотида включает регулирующие последовательности до и после раздела кодирования белка. Путем анализа этой последовательности можно определить нуклеотиды что код для аминокислот в итоговом белке.
После того, как у вас есть список генов, вы интересуетесь изучением, можно определить последовательности кодирования белка. Эта процедура использует человеческий ген HEXA и ген мыши HEXA как пример.
Если вы не получали генные данные с сети, можно загрузить данные в качестве примера из MAT-файла, включенного с программным обеспечением Bioinformatics Toolbox™. В Командном Окне MATLAB введите
load hexosaminidase
Структуры humanHEXA и mouseHEXA загрузите в рабочее пространство MATLAB.
Найдите открытые рамки считывания (ORFs) в человеческом гене. Например, для человеческого гена HEXA, ввести
humanORFs = seqshoworfs(humanHEXA.Sequence)
seqshoworfs создает структуру output humanORFs. Эта структура содержит положение запуска и кодонов остановки для всех открытых рамок считывания (ORFs) на каждой рамке считывания.
humanORFs =
1x3 struct array with fields:
Start
Stop
Браузер документации открывает отображение этих трех рамок считывания с ORFs, окрашенным в синий, красный цвет, и зеленый. Заметьте, что самый длинный ORF находится в системе координат первого чтения.

Найдите открытые рамки считывания (ORFs) в гене мыши. Ввод:
mouseORFs = seqshoworfs(mouseHEXA.Sequence)
seqshoworfs создает структуру mouseORFS.
mouseORFs =
1x3 struct array with fields:
Start
Stop
Ген мыши показывает самый длинный ORF на системе координат первого чтения.

Следующая процедура иллюстрирует, как использовать глобальные и локальные функции выравнивания, чтобы сравнить две последовательности аминокислот. Вы могли использовать функции выравнивания, чтобы искать общие черты между двумя последовательностями нуклеотида, но функции выравнивания возвращают более биологически значимые результаты, когда вы используете последовательности аминокислот.
После того, как вы определили местоположение открытых рамок считывания на своих последовательностях нуклеотида, можно преобразовать разделы кодирования белка последовательностей нуклеотида к их соответствующим последовательностям аминокислот, и затем можно сравнить их для общих черт.
Используя открытые рамки считывания, идентифицированные ранее, преобразуйте человека и последовательности DNA мыши к последовательностям аминокислот. Поскольку и человек и мышь, гены HEXA были в системах координат первого чтения (значение по умолчанию), вы не должны указывать который система координат. Ввод
humanProtein = nt2aa(humanHEXA.Sequence); mouseProtein = nt2aa(mouseHEXA.Sequence);
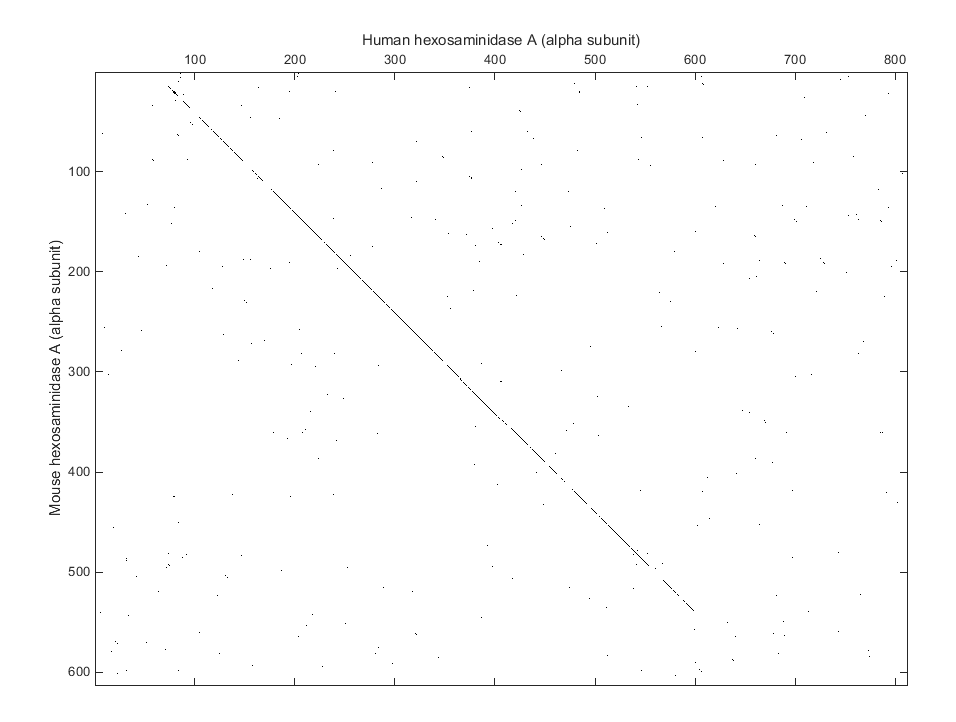
Постройте точечный график, сравнивающий последовательности аминокислот мыши и человек. Ввод
seqdotplot(mouseProtein,humanProtein,4,3)
ylabel('Mouse hexosaminidase A (alpha subunit)')
xlabel('Human hexosaminidase A (alpha subunit)')
Точечные диаграммы являются одним из самых легких способов искать подобие между последовательностями. Диагональная линия, показанная ниже, указывает, что может быть хорошее выравнивание между этими двумя последовательностями.
Глобально выровняйте эти две последовательности аминокислот, с помощью алгоритма Needleman-Wunsch. Ввод
[GlobalScore, GlobalAlignment] = nwalign(humanProtein,...
mouseProtein);
showalignment(GlobalAlignment)
showalignment отображает глобальное выравнивание этих двух последовательностей в Браузере документации. Заметьте, что расчетной идентичностью между этими двумя последовательностями является 60%.

Выравнивание очень хорошо между положением 69 и 599 аминокислоты, после которого эти две последовательности, кажется, не связаны. Заметьте, что существует остановка (*) в последовательности в этой точке. Если вы сокращаете последовательности, чтобы включать только аминокислоты, которые находятся в белке, вы можете получить лучшее выравнивание. Включайте положения аминокислоты от первого метионина (M) к первой остановке (*) это происходит после первого метионина.
Обрежьте последовательность, сначала запускают аминокислоту (обычно M) к первой остановке (*) и затем попробуйте выравнивание снова. Найдите индексы для остановок в последовательностях.
humanStops = find(humanProtein == '*')
humanStops =
41 599 611 713 722 730
mouseStops = find(mouseProtein == '*')
mouseStops =
539 557 574 606
Рассмотрение последовательности аминокислот для humanProtein, первый M в положении 70 и первой остановке после того, как то положение будет на самом деле второй остановкой в последовательности (положение 599). Рассмотрение последовательности аминокислот для mouseProtein, первый M в положении 11 и первой остановке после того, как то положение будет первой остановкой в последовательности (положение 557).
Обрежьте последовательности, чтобы включать только аминокислоты в белок и остановку.
humanProteinORF = humanProtein(70:humanStops(2)) humanProteinORF = MTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYVLYPNNFQFQYDV SSAAQPGCSVLDEAFQRYRDLLFGSGSWPRPYLTGKRHTLEKNVLVVSVV TPGCNQLPTLESVENYTLTINDDQCLLLSETVWGALRGLETFSQLVWKSA EGTFFINKTEIEDFPRFPHRGLLLDTSRHYLPLSSILDTLDVMAYNKLNV FHWHLVDDPSFPYESFTFPELMRKGSYNPVTHIYTAQDVKEVIEYARLRG IRVLAEFDTPGHTLSWGPGIPGLLTPCYSGSEPSGTFGPVNPSLNNTYEF MSTFFLEVSSVFPDFYLHLGGDEVDFTCWKSNPEIQDFMRKKGFGEDFKQ LESFYIQTLLDIVSSYGKGYVVWQEVFDNKVKIQPDTIIQVWREDIPVNY MKELELVTKAGFRALLSAPWYLNRISYGPDWKDFYIVEPLAFEGTPEQKA LVIGGEACMWGEYVDNTNLVPRLWPRAGAVAERLWSNKLTSDLTFAYERL SHFRCELLRRGVQAQPLNVGFCEQEFEQT* mouseProteinORF = mouseProtein(11:mouseStops(1)) mouseProteinORF = MAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYTLYPNNFQFRYHV SSAAQAGCVVLDEAFRRYRNLLFGSGSWPRPSFSNKQQTLGKNILVVSVV TAECNEFPNLESVENYTLTINDDQCLLASETVWGALRGLETFSQLVWKSA EGTFFINKTKIKDFPRFPHRGVLLDTSRHYLPLSSILDTLDVMAYNKFNV FHWHLVDDSSFPYESFTFPELTRKGSFNPVTHIYTAQDVKEVIEYARLRG IRVLAEFDTPGHTLSWGPGAPGLLTPCYSGSHLSGTFGPVNPSLNSTYDF MSTLFLEISSVFPDFYLHLGGDEVDFTCWKSNPNIQAFMKKKGFTDFKQL ESFYIQTLLDIVSDYDKGYVVWQEVFDNKVKVRPDTIIQVWREEMPVEYM LEMQDITRAGFRALLSAPWYLNRVKYGPDWKDMYKVEPLAFHGTPEQKAL VIGGEACMWGEYVDSTNLVPRLWPRAGAVAERLWSSNLTTNIDFAFKRLS HFRCELVRRGIQAQPISVGCCEQEFEQT*
Глобально выровняйте обрезанные последовательности аминокислот. Ввод
[GlobalScore_trim, GlobalAlignment_trim] = nwalign(humanProteinORF,...
mouseProteinORF);
showalignment(GlobalAlignment_trim)
showalignment отображает результаты для второго глобального выравнивания. Заметьте, что идентичностью процента для необрезанных последовательностей является 60% и 84% для обрезанных последовательностей.

Другой способ обрезать последовательность аминокислот только до тех аминокислот в белке состоит в том, чтобы сначала обрезать последовательность нуклеотида с индексами от seqshoworfs функция. Помните, что ORF для человеческого гена HEXA и ORF для мыши HEXA были оба на системе координат первого чтения.
humanORFs = seqshoworfs(humanHEXA.Sequence)
humanORFs =
1x3 struct array with fields:
Start
Stop
mouseORFs = seqshoworfs(mouseHEXA.Sequence)
mouseORFs =
1x3 struct array with fields:
Start
Stop
humanPORF = nt2aa(humanHEXA.Sequence(humanORFs(1).Start(1):...
humanORFs(1).Stop(1)));
mousePORF = nt2aa(mouseHEXA.Sequence(mouseORFs(1).Start(1):...
mouseORFs(1).Stop(1)));
[GlobalScore2, GlobalAlignment2] = nwalign(humanPORF, mousePORF);
Покажите выравнивание в Браузере документации.
showalignment(GlobalAlignment2)
Результат сначала обрезающий последовательность нуклеотида прежде, чем преобразовать это в последовательность аминокислот совпадает с результатом усечения последовательности аминокислот после преобразования. Смотрите результат на шаге 6.
Альтернативный метод к работе с подпоследовательностями должен использовать локальную функцию выравнивания с неусеченными последовательностями.
Локально выровняйте эти две последовательности аминокислот с помощью алгоритма Смита-лодочника. Ввод
[LocalScore, LocalAlignment] = swalign(humanProtein,...
mouseProtein)
LocalScore =
1057
LocalAlignment =
RGDQR-AMTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYV . . .
|| | ||:: ||| |||||||:| ||||||||| :|| :||: . . .
RGAGRWAMAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYT . . .
Покажите выравнивание в цвете.
showalignment(LocalAlignment)

