Выровняйте массовые спектры из нескольких пиковых списков от набора данных GC/MS или LC/MS
[CMZ, AlignedPeaks]
= mspalign(Peaklist)
[CMZ, AlignedPeaks]
= mspalign(Peaklist, ...'Quantile', QuantileValue,
...)
[CMZ, AlignedPeaks]
= mspalign(Peaklist, ...'EstimationMethod', EstimationMethodValue,
...)
[CMZ, AlignedPeaks]
= mspalign(Peaklist, ...'CorrectionMethod', CorrectionMethodValue,
...)
[CMZ, AlignedPeaks]
= mspalign(Peaklist, ...'ShowEstimation', ShowEstimationValue,
...)
Peaklist | Массив ячеек пиковых списков от жидкостной хроматографии / масс-спектрометрия (LC/MS) или газовая хроматография/масс-спектрометрия (GC/MS) набор данных. Каждый элемент в массиве ячеек является матрицей 2D столбца с m/z значениями в первом столбце и ионными значениями интенсивности во втором столбце. Каждый элемент соответствует время задержания или спектру.ПримечаниеМожно использовать функцию |
QuantileValue | Значение, которое определяет, какой peaks выбран методом оценки создать CMZ, вектор общих m/z значений. Выбором является любое значение ≥ 0 и ≤ 1. Значением по умолчанию является 0.95. |
EstimationMethodValue | Вектор символов или строка, задающая метод, чтобы оценить CMZ, вектор общей массы/заряда (m/z) значения. Выбор:
|
CorrectionMethodValue | Вектор символов или строка, задающая метод, чтобы выровнять каждый пиковый список к вектору CMZ. Выбор:
|
ShowEstimationValue | Управляет отображением графика оценки относительно метода оценки и вектора общей массы/заряда (m/z) значения. Выбором является true или false. Значение по умолчанию также:
|
CMZ | Вектор общей массы/заряда (m/z) значения оценивается функцией mspalign. |
AlignedPeaks | Массив ячеек пиковых списков, с той же формой как Peaklist, но с исправленными m/z значениями в первом столбце каждой матрицы. |
[ выравнивает массовые спектры из нескольких пиковых списков (центроидные данные), первой оценкой CMZ, AlignedPeaks]
= mspalign(Peaklist)CMZ, вектор общей массы/заряда (m/z) значения, оцененные путем рассмотрения peaks во всех спектрах в Peaklist, массиве ячеек пиковых списков, где каждый элемент соответствует время задержания или спектру. Это затем выравнивает peaks в каждом спектре к значениям в CMZ, создавая AlignedPeaks, массив ячеек выровненных пиковых списков.
[CMZ, AlignedPeaks] = mspalign(Peaklist, ...'PropertyName', PropertyValue, ...) mspalign с дополнительными свойствами, которые используют имя свойства / пары значения свойства. Можно задать одно или несколько свойств в любом порядке. Каждый PropertyName должен быть заключен в одинарные кавычки и нечувствительный к регистру. Это имя свойства / пары значения свойства следующие:
[ определяет, какой peaks выбран методом оценки создать CMZ, AlignedPeaks]
= mspalign(Peaklist, ...'Quantile', QuantileValue,
...)CMZ, вектор общих m/z значений. Выбором является скаляр между 0 и 1. Значением по умолчанию является 0.95.
[ указывает, что метод раньше оценивал CMZ, AlignedPeaks]
= mspalign(Peaklist, ...'EstimationMethod', EstimationMethodValue,
...)CMZ, вектор общей массы/заряда (m/z) значения. Выбор:
гистограмма Метод по умолчанию. Пиковые местоположения кластеризируются с помощью подхода оценки плотности ядра. Пиковая ионная интенсивность используется в качестве фактора взвешивания. Центр всех кластеров соответствует вектору CMZ.
regression — Берет выборку расстояний между наблюдаемым значительным peaks и регрессами межпиковое расстояние, чтобы создать вектор CMZ с подобными межэлементными расстояниями.
[ указывает, что метод раньше выравнивал каждый пиковый список к вектору CMZ, AlignedPeaks]
= mspalign(Peaklist, ...'CorrectionMethod', CorrectionMethodValue,
...)CMZ. Выбор:
nearestNeighbor Метод по умолчанию. Для каждого общего пика в векторе CMZ его дубликат в каждом пиковом списке является пиком, который является самым близким к m/z значению общего пика.
кратчайший путь Для каждого общего пика в векторе CMZ его дубликат в каждом пиковом списке выбран с помощью алгоритма поиска кратчайшего пути.
[ управляет отображением графика оценки относительно метода оценки и предполагаемого вектора общей массы/заряда (m/z) значения. Выбором является CMZ, AlignedPeaks]
= mspalign(Peaklist, ...'ShowEstimation', ShowEstimationValue,
...)true или false. Значение по умолчанию также:
ложь Когда возвращаемые значения заданы.
tRUE Когда возвращаемые значения не заданы.
Загрузите MAT-файл, включенный с программным обеспечением Bioinformatics Toolbox™, которое содержит жидкостную хроматографию / переменные данных (LC/MS) масс-спектрометрии, включая peaks и ret_time. peaks является массивом ячеек пиковых списков, где каждый элемент является матрицей 2D столбца m/z значений и ионных значений интенсивности, и каждый элемент соответствует время задержания или спектру. ret_time является вектор-столбцом времен задержания, сопоставленных с набором данных LC/MS.
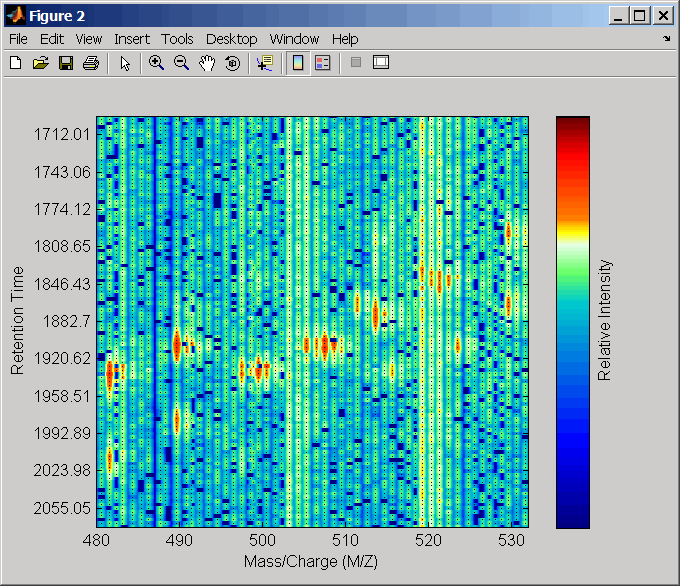
load lcmsdataПередискретизируйте невыровненные данные, отобразите их в карте тепла, и затем наложите точечную диаграмму.
[MZ,Y] = msppresample(ms_peaks,5000); msheatmap(MZ,ret_time,log(Y))
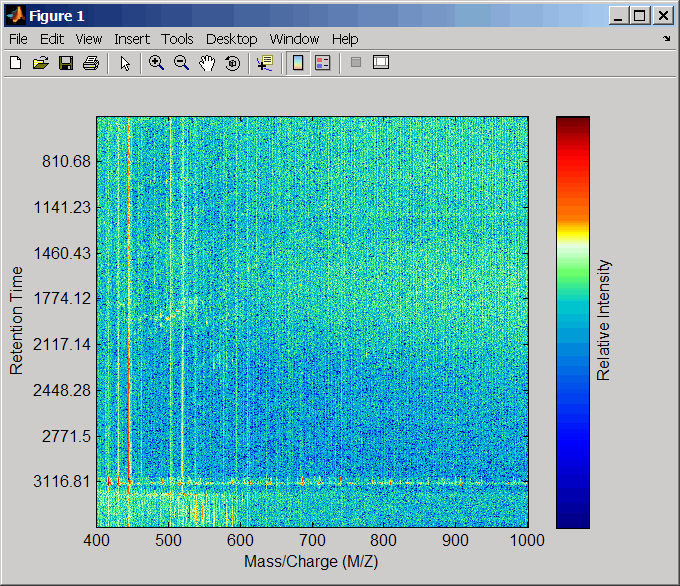
msdotplot(ms_peaks,ret_time)
Нажмите Zoom в![]() кнопке, и затем кликните по точечной диаграмме два или три раза, чтобы увеличить масштаб и видеть, как точки, представляющие peaks, накладывают изображение карты тепла.
кнопке, и затем кликните по точечной диаграмме два или три раза, чтобы увеличить масштаб и видеть, как точки, представляющие peaks, накладывают изображение карты тепла.
Выровняйте пиковые списки от массовых спектров с помощью методов оценки и исправления по умолчанию.
[CMZ, aligned_peaks] = mspalign(ms_peaks);
Передискретизируйте невыровненные данные, отобразите их в карте тепла, и затем наложите точечную диаграмму.
[MZ2,Y2] = msppresample(aligned_peaks,5000); msheatmap(MZ2,ret_time,log(Y2))
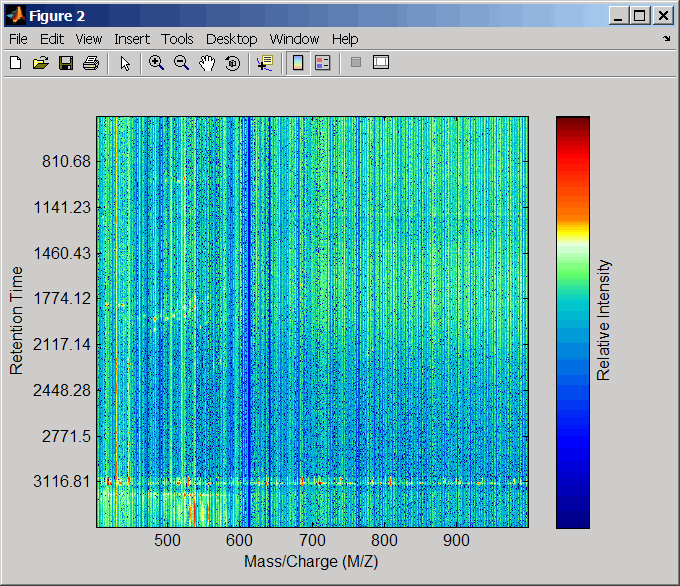
msdotplot(aligned_peaks,ret_time)
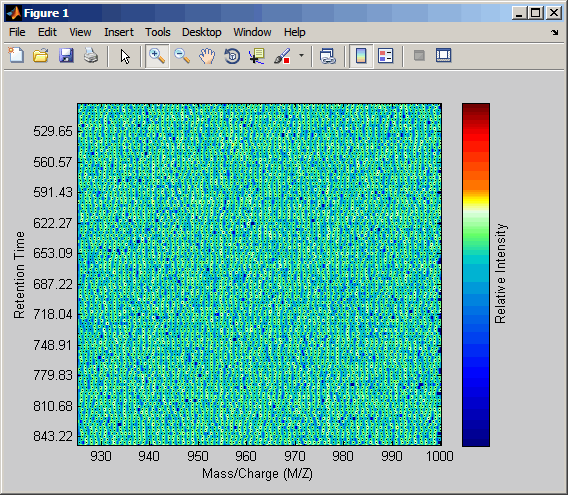
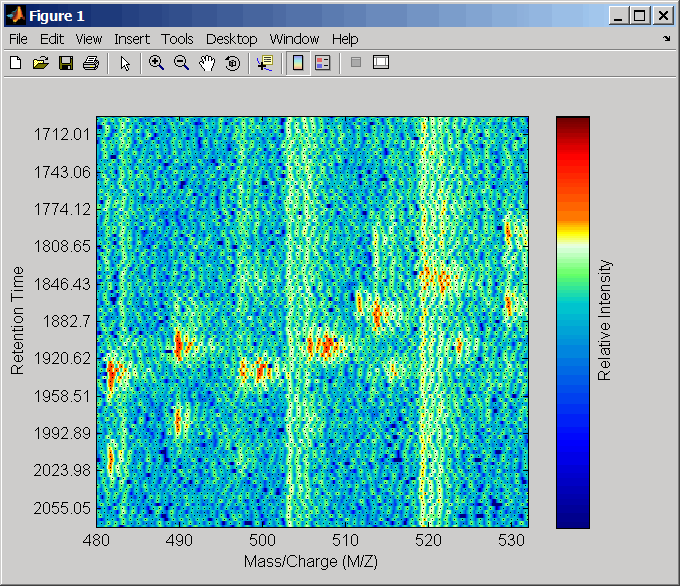
Соедините оси двух графиков тепла и увеличения, чтобы наблюдать, что деталь сравнивает невыровненные и выровненные наборы данных LC/MS.
linkaxes(findobj(0,'Tag','MSHeatMap')) axis([480 532 375 485])
[1] Jeffries, N. (2005) Алгоритмы для выравнивания масс-спектрометрии протеомные данные. Bioinfomatics 21:14, 3066–3073.
[2] Purvine, S., Kolker, N. и Kolker, E. (2004) спектральная качественная оценка для тандемной протеомики масс-спектрометрии Высокой Пропускной способности. OMICS: журнал интегральной биологии 8:3, 255–265.
msalign | msdotplot | msheatmap | mspeaks | msppresample | mzcdf2peaks | mzxml2peaks
