Локально выровняйте две последовательности с помощью алгоритма Смита-лодочника
Score = swalign(Seq1, Seq2)
[Score, Alignment] = swalign(Seq1, Seq2)
[Score, Alignment, Start]
= swalign(Seq1, Seq2)
... = swalign(Seq1,Seq2,
...'Alphabet', AlphabetValue)
... = swalign(Seq1,Seq2,
...'ScoringMatrix', ScoringMatrixValue,
...)
... = swalign(Seq1,Seq2,
...'Scale', ScaleValue, ...)
... = swalign(Seq1,Seq2,
...'GapOpen', GapOpenValue, ...)
... = swalign(Seq1,Seq2,
...'ExtendGap', ExtendGapValue, ...)
... = swalign(Seq1,Seq2,
...'Showscore', ShowscoreValue, ...)
Seq1, Seq2 | Аминокислота или последовательности нуклеотида. Введите любое следующее: Совет Для справки с буквой и целочисленными представлениями аминокислот и нуклеотидов, смотрите Поиск Поиска или Нуклеотида Аминокислоты. |
AlphabetValue | Вектор символов или строка, задающая тип последовательности. Выбором является 'AA' (значение по умолчанию) или 'NT'. |
ScoringMatrixValue | Любое из следующего:
Примечание Если необходимо скомпилировать |
ScaleValue | Положительное значение, которое задает масштабный коэффициент, который применяется к выходному счету. Например, если выходной счет первоначально определяется в битах, и вы вводите Значением по умолчанию является Примечание Если Совет Прежде, чем сравнить баллы выравнивания от нескольких выравниваний, гарантируйте, что баллы находятся в тех же модулях. Можно использовать |
GapOpenValue | Положительное значение, задающее штраф за открытие разрыва в выравнивании. Значением по умолчанию является |
ExtendGapValue | Положительное значение, задающее штраф за расширение разрыва с помощью аффинной схемы штрафа разрыва. Примечание Если вы задаете это значение, |
ShowscoreValue | Управляет отображением пробела выигрыша и путем к победе выравнивания. Выбором является true или false (значение по умолчанию). |
Score | Оптимальное локальное выравнивание выигрывает в битах. |
Alignment | 3 N символьным массивом, показывающим эти две последовательности, Seq1 и Seq2, в первых и третьих строках и символах, представляющих оптимальное локальное выравнивание между ними во второй строке. |
Start | 2 1 вектор из индексов, указывающих на начальную точку в каждой последовательности для выравнивания. |
Score = swalign(Seq1, Seq2)
[ возвращает 3 N символьным массивом, показывающим эти две последовательности, Score, Alignment] = swalign(Seq1, Seq2)Seq1 и Seq2, в первых и третьих строках и символах, представляющих оптимальное локальное выравнивание между ними во второй строке. Символ | указывает на аминокислоты или нуклеотиды то соответствие точно. Символ : указывает на аминокислоты или нуклеотиды, которые связаны, как задано матрицей выигрыша (несовпадения с нулем или положительным выигрывающим матричным значением).
[ возвращается 2 1 вектор из индексов, указывающих на начальную точку в каждой последовательности для выравнивания.Score, Alignment, Start]
= swalign(Seq1, Seq2)
... = swalign ( вызовы Seq1, Seq2PropertyName ', PropertyValue, ...)swalign с дополнительными свойствами, которые используют имя свойства / пары значения свойства. Можно задать одно или несколько свойств в любом порядке. Каждый PropertyName должен быть заключен в одинарные кавычки и нечувствительный к регистру. Это имя свойства / пары значения свойства следующие:
... = swalign( задает тип последовательностей. Выбором является Seq1,Seq2,
...'Alphabet', AlphabetValue)'AA' (значение по умолчанию) или 'NT'.
... = swalign( задает матрицу выигрыша, чтобы использовать для локального выравнивания. Значение по умолчанию:Seq1,Seq2,
...'ScoringMatrix', ScoringMatrixValue,
...)
'BLOSUM50' — Когда AlphabetValue равняется 'AA'
'NUC44' — Когда AlphabetValue равняется 'NT'
... = swalign( задает масштабный коэффициент, который применяется к выходному счету, таким образом, управляя модулями выходного счета. Выбором является любое положительное значение.Seq1,Seq2,
...'Scale', ScaleValue, ...)
... = swalign( задает штраф за открытие разрыва в выравнивании. Выбором является любое положительное значение. Значением по умолчанию является Seq1,Seq2,
...'GapOpen', GapOpenValue, ...)8.
... = swalign( задает штраф за расширение разрыва с помощью аффинной схемы штрафа разрыва. Выбором является любое положительное значение. Seq1,Seq2,
...'ExtendGap', ExtendGapValue, ...)
... = swalign( управляет отображением пробела выигрыша и завоевания пути выравнивания. Выбором является Seq1,Seq2,
...'Showscore', ShowscoreValue, ...)true или false (значение по умолчанию).
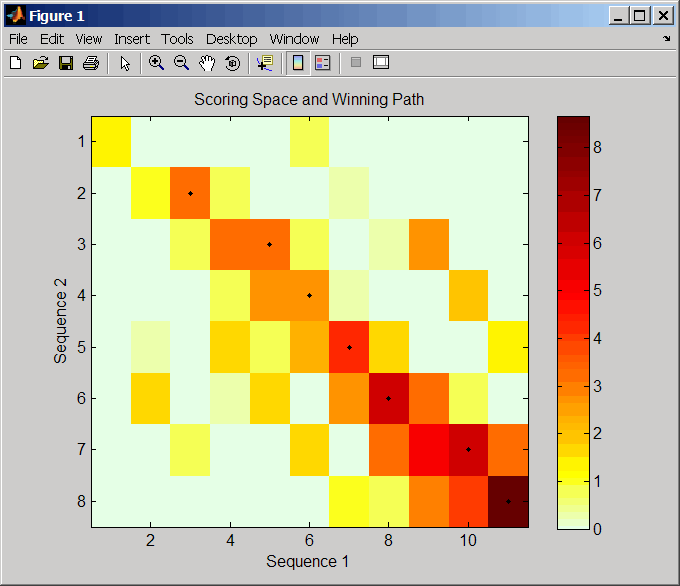
Пробел выигрыша является картой тепла, отображающей лучшую музыку ко всем частичным выравниваниям двух последовательностей. Цвет каждого (n1,n2) координата на пробеле выигрыша представляет лучший счет к соединению подпоследовательностей Seq1(s1:n1) и Seq2(s2:n2), где n1 положение в Seq1, n2 положение in Seq2S1 любое положение в Seq1 между 1:n1, и s2 любое положение в Seq2 между 1:n2. Лучший счет к соединению определенных подпоследовательностей определяется путем выигрыша всех возможных выравниваний подпоследовательностей путем подведения итогов штрафов разрыва и соответствий.
Путь к победе представлен черными точками на пробеле выигрыша, и это иллюстрирует соединение положений в оптимальном локальном выравнивании. Цвет последней точки (нижний правый угол) пути к победе представляет оптимальный локальный счет выравнивания к этим двум последовательностям и является Score выведите возвращенный swalign.
Примечание
Пробел выигрыша визуально показывает тандемные повторения, маленькие сегменты, которые потенциально выравниваются, и частичные выравнивания областей от перестроенных последовательностей.
Локально выровняйте две последовательности аминокислот с помощью BLOSUM50 (значение по умолчанию) матрица выигрыша и значения по умолчанию для GapOpen и ExtendGap свойства. Возвратите оптимальный локальный счет выравнивания в битах и символьном массиве выравнивания.
[Score, Alignment] = swalign('VSPAGMASGYD','IPGKASYD') Score = 8.6667 Alignment = PAGMASGYD | | || || P-GKAS-YD
Локально выровняйте две последовательности аминокислот, задающие PAM250 выигрыш матрицы и разрыва открывает штраф 5.
[Score, Alignment] = swalign('HEAGAWGHEE','PAWHEAE',... 'ScoringMatrix', 'pam250',... 'GapOpen',5) Score = 8 Alignment = GAWGHE :|| || PAW-HE
Локально выровняйте две последовательности аминокислот, возвращающие Score в туземных модулях (nats) путем определения масштабного коэффициента log(2).
[Score, Alignment] = swalign('HEAGAWGHEE','PAWHEAE','Scale',log(2)) Score = 6.4694 Alignment = AWGHE || || AW-HE
[1] Durbin, R., вихрь, S., Krogh, A. и Мичисон, G. (1998). Биологический анализ последовательности (издательство Кембриджского университета).
[2] Смит, T., и Лодочник, М. (1981). Идентификация общих молекулярных подпоследовательностей. Журнал Молекулярной биологии 147, 195–197.
aa2int | aminolookup | baselookup | blosum | dayhoff | gonnet | int2aa | int2nt | localalign | multialign | nt2aa | nt2int | nuc44 | nwalign | pam | pdbsuperpose | seqdotplot
