Выравнивание масс-спектров из нескольких пиковых списков из набора данных LC/MS или GC/MS
[CMZ, AlignedPeaks] = mspalign(Peaklist)
[CMZ, AlignedPeaks] = mspalign(Peaklist, ...'Quantile', QuantileValue, ...)
[CMZ, AlignedPeaks] = mspalign(Peaklist, ...'EstimationMethod', EstimationMethodValue, ...)
[CMZ, AlignedPeaks] = mspalign(Peaklist, ...'CorrectionMethod', CorrectionMethodValue, ...)
[CMZ, AlignedPeaks] = mspalign(Peaklist, ...'ShowEstimation', ShowEstimationValue, ...)
Peaklist | Клеточный массив пиковых списков из набора данных жидкостной хроматографии/масс-спектрометрии (ЖХ/МС) или газовой хроматографии/масс-спектрометрии (ГХ/МС). Каждый элемент в массиве ячеек представляет собой матрицу из двух столбцов со значениями m/z в первом столбце и значениями интенсивности ионов во втором столбце. Каждый элемент соответствует спектру или времени удерживания. Примечание Вы можете использовать |
QuantileValue | Значение, определяющее, какие пики выбираются методом оценки для создания CMZ, вектор общих значений m/z. Варианты - это любое значение ≥ 0 и ≤ 1. По умолчанию: 0.95. |
EstimationMethodValue | Символьный вектор или строка, задающая метод оценки CMZ, вектор общих значений масса/заряд (m/z). Возможны следующие варианты:
|
CorrectionMethodValue | Символьный вектор или строка, задающая метод выравнивания каждого списка пиков по CMZ вектор. Возможны следующие варианты:
|
ShowEstimationValue | Управляет отображением графика оценки относительно метода оценки и вектора общих значений массы/заряда (m/z). Варианты: true или false. Значение по умолчанию:
|
CMZ | Вектор значений общей массы/заряда (m/z), оцененных по mspalign функция. |
AlignedPeaks | Массив ячеек списков пиков с той же формой, что и Peaklist, но с скорректированными значениями m/z в первом столбце каждой матрицы. |
[ выравнивает масс-спектры из нескольких пиковых списков (центроидализированные данные) путем первой оценки CMZ, AlignedPeaks] = mspalign(Peaklist)CMZвектор общих значений масса/заряд (m/z), оцененный путем учета пиков во всех спектрах в Peaklistмассив ячеек списков пиков, где каждый элемент соответствует спектру или времени удержания. Затем он выравнивает пики в каждом спектре по значениям в CMZ, создание AlignedPeaksмассив ячеек выровненных списков пиков.
[ требования CMZ, AlignedPeaks] = mspalign(Peaklist, ...'PropertyName', PropertyValue, ...)mspalign с необязательными свойствами, использующими пары имя/значение свойства. Можно указать одно или несколько свойств в любом порядке. Каждый PropertyName должен быть заключен в одинарные кавычки и не учитывать регистр. Эти пары имя/значение свойства следующие:
[ определяет, какие пики выбираются методом оценки для создания CMZ, AlignedPeaks] = mspalign(Peaklist, ...'Quantile', QuantileValue, ...)CMZ, вектор общих значений m/z. Варианты являются скаляром между 0 и 1. По умолчанию: 0.95.
[ указывает метод, используемый для оценки CMZ, AlignedPeaks] = mspalign(Peaklist, ...'EstimationMethod', EstimationMethodValue, ...)CMZ, вектор общих значений масса/заряд (m/z). Возможны следующие варианты:
histogram - Метод по умолчанию. Местоположения пиков группируются с использованием подхода оценки плотности ядра. Пиковая интенсивность ионов используется в качестве весового коэффициента. Центр всех кластеров соответствует CMZ вектор.
regression - Берет выборку расстояний между наблюдаемыми значительными пиками и регрессирует межпиковое расстояние для создания CMZ вектор с аналогичными межэлементными расстояниями.
[ задает метод, используемый для выравнивания каждого списка пиков с CMZ, AlignedPeaks] = mspalign(Peaklist, ...'CorrectionMethod', CorrectionMethodValue, ...)CMZ вектор. Возможны следующие варианты:
nearest-neighbor - Метод по умолчанию. Для каждого общего пика в CMZ вектор, его аналог в каждом списке пиков является пиком, который ближе всего к значению m/z общего пика.
shortest-path - Для каждого общего пика в CMZ вектор, его аналог в каждом списке пиков выбирается с использованием алгоритма кратчайшего пути.
[ управляет отображением графика оценки относительно метода оценки и оценочным вектором значений общей массы/заряда (m/z). Варианты: CMZ, AlignedPeaks] = mspalign(Peaklist, ...'ShowEstimation', ShowEstimationValue, ...)true или false. Значение по умолчанию:
false - Когда указаны возвращаемые значения.
true - Когда возвращаемые значения не указаны.
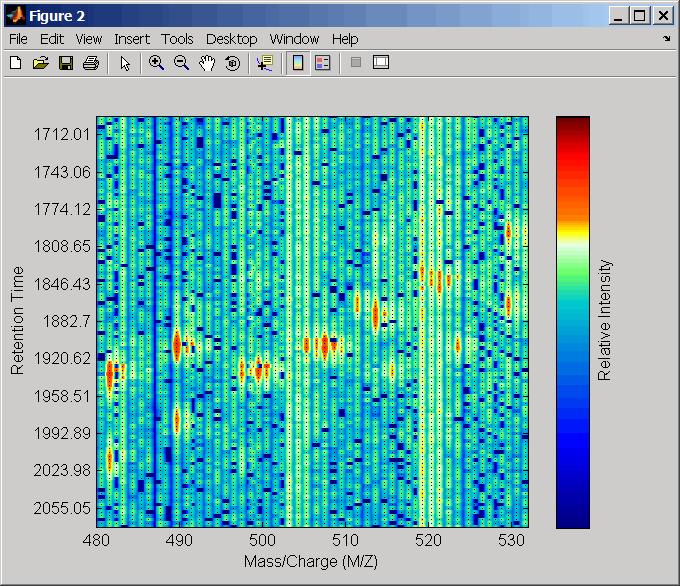
Загрузите MAT-файл, включенный в программное обеспечение Bioinformatics Toolbox™, который содержит переменные данных жидкостной хроматографии/масс-спектрометрии (LC/MS), включая peaks и ret_time. peaks является массивом ячеек пиковых списков, где каждый элемент является матрицей из двух столбцов значений m/z и значений интенсивности ионов, и каждый элемент соответствует спектру или времени удержания. ret_time - вектор столбца времени хранения, связанный с набором данных LC/MS.
load lcmsdataВыполните повторную выборку неориентированных данных, отобразите их на тепловой карте, а затем наложите точечный график.
[MZ,Y] = msppresample(ms_peaks,5000); msheatmap(MZ,ret_time,log(Y))
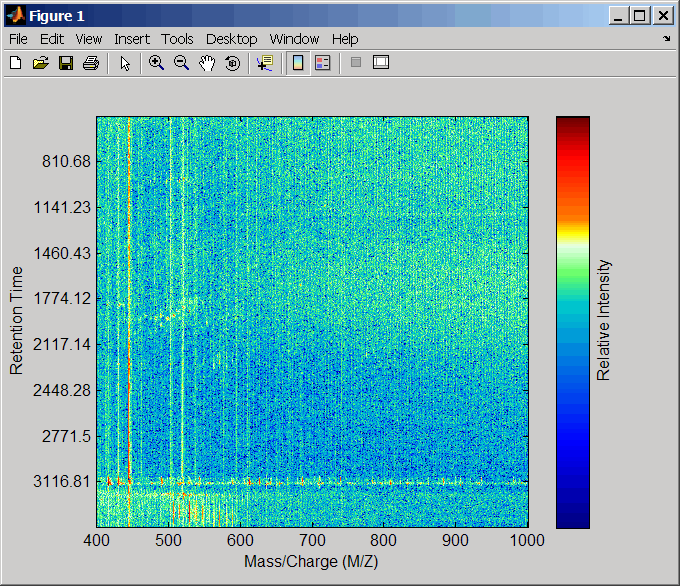
msdotplot(ms_peaks,ret_time)
Нажмите![]() кнопку «Увеличить», а затем два или три раза щелкните точечный график, чтобы увеличить масштаб изображения и увидеть, как точки, представляющие пики, накладываются на изображение тепловой карты.
кнопку «Увеличить», а затем два или три раза щелкните точечный график, чтобы увеличить масштаб изображения и увидеть, как точки, представляющие пики, накладываются на изображение тепловой карты.
Выровняйте списки пиков из масс-спектров с помощью методов оценки и коррекции по умолчанию.
[CMZ, aligned_peaks] = mspalign(ms_peaks);
Выполните повторную выборку неориентированных данных, отобразите их на тепловой карте, а затем наложите точечный график.
[MZ2,Y2] = msppresample(aligned_peaks,5000); msheatmap(MZ2,ret_time,log(Y2))
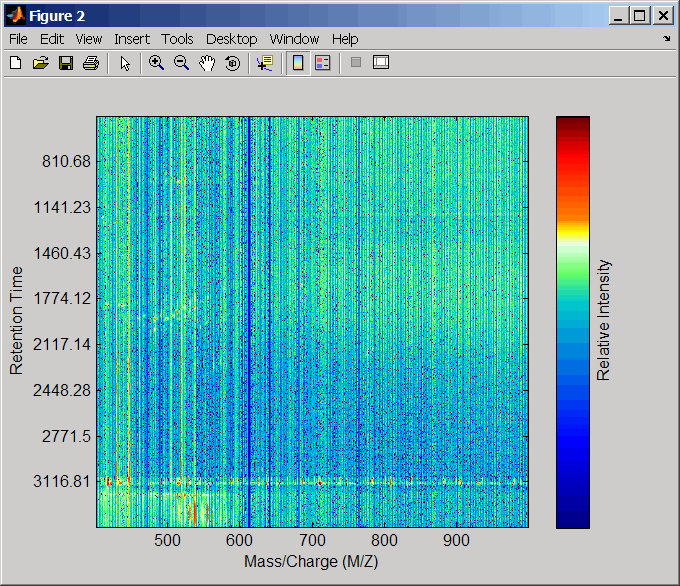
msdotplot(aligned_peaks,ret_time)
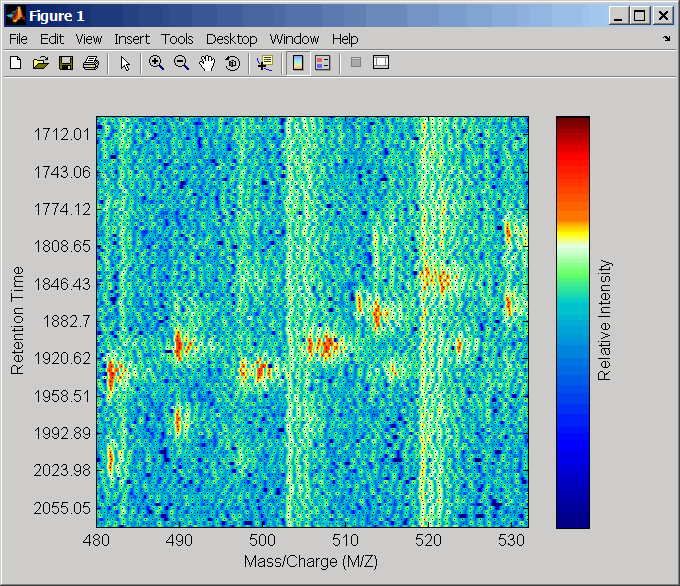
Свяжите оси двух тепловых графиков и увеличьте масштаб изображения, чтобы увидеть детали для сравнения несцентрированных и выровненных наборов данных LC/MS.
linkaxes(findobj(0,'Tag','MSHeatMap')) axis([480 532 375 485])
[1] Джеффрис, Н. (2005) Алгоритмы выравнивания протеомных данных масс-спектрометрии. Биоинфоматика 21:14, 3066-3073.
[2] Purvine, S., Kolker, N. и Kolker, E. (2004) Оценка спектрального качества для высокопрочной тандемной масс-спектрометрии протеомики. OMICS: Журнал интегративной биологии 8:3, 255-265.
msalign | msbackadj | msdotplot | msheatmap | mslowess | msnorm | mspalign | mspeaks | msppresample | msresample | mssgolay | msviewer
