Проверка кластеров в филогенетическом дереве
LeafClusters = cluster(Tree, Threshold)
[LeafClusters, NodeClusters] = cluster(Tree, Threshold)
[LeafClusters, NodeClusters, Branches] = cluster(Tree, Threshold)
cluster(..., 'Criterion', CriterionValue, ...)
cluster(..., 'MaxClust', MaxClustValue, ...)
cluster(..., 'Distances', DistancesValue, ...)
Tree | Созданный объект филогенетического дерева, например, созданный с помощью |
Threshold | Скаляр, указывающий пороговое значение. |
CriterionValue | Символьный вектор или строка, задающая критерий для определения количества кластеров как функции парных расстояний видов. Возможны следующие варианты:
|
MaxClustValue | Положительное целое число, указывающее максимальное количество возможных кластеров для проверяемых разделов. По умолчанию - количество листьев в дереве. Совет При использовании Совет При использовании |
DistancesValue | Матрица попарных расстояний, например, возвращаемых |
LeafClusters | Вектор столбца, содержащий индекс кластера для каждого вида (листа) в |
NodeClusters | Вектор столбца, содержащий индекс кластера для каждого конечного узла и узла ветви в |
Branches | Матрица из двух столбцов, содержащая для каждого шага алгоритма индекс рассматриваемой ветви и значение критерия. Каждая строка соответствует шагу алгоритма. Первый столбец содержит индексы ветвей, а второй столбец содержит значения критериев. Совет Для получения всей кривой критерия в зависимости от количества кластеров в |
LeafClusters = cluster(Tree, Threshold)
Начиная с двух кластеров (k = 2), выбирает раздел, оптимизирующий критерий, заданный 'Criterion' собственность
Приращения k на 1 и снова выбирает оптимальный раздел
Продолжает приращение k и выбор оптимального раздела до значения критерия = Threshold или k = максимальное количество скоплений (то есть количество листьев)
Из всех возможных значений k выбирает значение k, разбиение которого оптимизирует критерий
[ возвращает вектор столбца, содержащий индекс кластера для каждого конечного узла и узла ветви в LeafClusters, NodeClusters] = cluster(Tree, Threshold)Tree.
[ возвращает матрицу из двух столбцов, содержащую для каждого шага алгоритма индекс рассматриваемой ветви и значение критерия. Каждая строка соответствует шагу алгоритма. Первый столбец содержит индексы ветвей, а второй столбец содержит значения критериев.LeafClusters, NodeClusters, Branches] = cluster(Tree, Threshold)
cluster(..., ' требования PropertyName', PropertyValue, ...)cluster с необязательными свойствами, использующими пары имя/значение свойства. Можно указать одно или несколько свойств в любом порядке. Заключить каждый PropertyName в одинарных кавычках. Каждый PropertyName нечувствителен к регистру. Эти пары имя/значение свойства следующие:.
cluster(..., 'Criterion', определяет критерий для определения количества кластеров как функции парных расстояний видов. CriterionValue, ...)
cluster(..., 'MaxClust', указывает максимальное количество возможных кластеров для тестируемых разделов. По умолчанию - количество листьев в дереве.MaxClustValue, ...)
cluster(..., 'Distances', заменяет патристические расстояния в DistancesValue, ...)Tree с предоставленной пользователем парной матрицей расстояний.
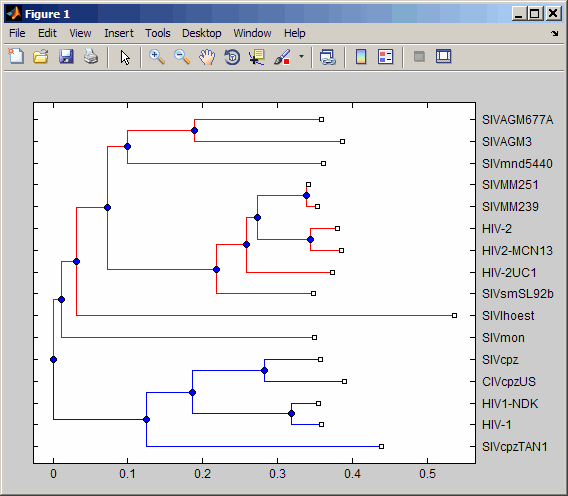
Проверка кластеров в филогенетическом дереве:
% Read sequences from a multiple alignment file into a MATLAB
% structure
gagaa = multialignread('aagag.aln');
% Build a phylogenetic tree from the sequences
gag_tree = seqneighjoin(seqpdist(gagaa),'equivar',gagaa);
% Validate the clusters in the tree and find the best partition
% using the 'gain' criterion
[i,j] = cluster(gag_tree,[],'criterion','gain','maxclust',10);
% Use the returned vector of indices to color the branches of each
% cluster in a plot of the tree
h = plot(gag_tree);
set(h.BranchLines(j==2),'Color','b')
set(h.BranchLines(j==1),'Color','r')
[1] Дудойт, С. и Фридлян, Дж. (2002). Способ повторной выборки на основе прогнозирования для оценки количества кластеров в наборе данных. Биология генома 3 (7), исследование 0036.1-0036.21.
[2] Теодоридис, С. и Коутрумбас, К. (1999). Распознавание образов (академическая пресса), стр. 434-435.
[3] Кауфман, Л. и Руссью, P.J. (1990). Поиск групп в данных: введение в кластерный анализ (Нью-Йорк, Уайли).
[4] Калински, Р. и Харабаш, Дж. (1974). Дендритный метод кластерного анализа. Commun Statistics 3, 1-27.
[5] Хартиган, J.A. (1985). Статистическая теория в кластеризации. J Классификация 2, 63-76.
cluster | phytree | phytreeread | phytreeviewer | plot | seqlinkage | seqneighjoin | seqpdist | silhouette | view
