Локальное выравнивание двух последовательностей с использованием алгоритма Смита-Уотермана
Score = swalign(Seq1, Seq2)
[Score, Alignment] = swalign(Seq1, Seq2)
[Score, Alignment, Start] = swalign(Seq1, Seq2)
... = swalign(Seq1,Seq2, ...'Alphabet', AlphabetValue)
... = swalign(Seq1,Seq2, ...'ScoringMatrix', ScoringMatrixValue, ...)
... = swalign(Seq1,Seq2, ...'Scale', ScaleValue, ...)
... = swalign(Seq1,Seq2, ...'GapOpen', GapOpenValue, ...)
... = swalign(Seq1,Seq2, ...'ExtendGap', ExtendGapValue, ...)
... = swalign(Seq1,Seq2, ...'Showscore', ShowscoreValue, ...)
Seq1, Seq2 | Аминокислотные или нуклеотидные последовательности. Введите любое из следующих значений: Совет Для получения справки по буквенным и целочисленным представлениям аминокислот и нуклеотидов см. Аминокислотный или нуклеотидный поиск. |
AlphabetValue | Символьный вектор или строка, указывающая тип последовательности. Варианты: 'AA' (по умолчанию) или 'NT'. |
ScoringMatrixValue | Одно из следующих действий:
Примечание Если нужно скомпилировать |
ScaleValue | Положительное значение, указывающее масштабный коэффициент, применяемый к выходной шкале. Например, если исходная оценка первоначально определяется в битах, и вводится значение По умолчанию: Примечание Если Совет Прежде чем сравнивать оценки выравнивания из нескольких трасс, убедитесь, что оценки находятся в одних и тех же единицах. Вы можете использовать |
GapOpenValue | Положительное значение, определяющее штраф за открытие зазора в трассе. По умолчанию: |
ExtendGapValue | Положительное значение, определяющее штраф за расширение разрыва с использованием аффинной схемы штрафов за разрыв. Примечание Если указано это значение, |
ShowscoreValue | Управление отображением пространства оценки и траектории выигрыша трассы. Варианты: true или false (по умолчанию). |
Score | Оптимальная оценка локального выравнивания в битах. |
Alignment | 3-by-N символьный массив, показывающий две последовательности, Seq1 и Seq2в первой и третьей строках и символы, представляющие оптимальное локальное выравнивание между ними во второй строке. |
Start | 2-на-1 вектор индексов, указывающий начальную точку в каждой последовательности для выравнивания. |
Score = swalign(Seq1, Seq2)
[ возвращает 3-by-N символьный массив, показывающий две последовательности, Score, Alignment] = swalign(Seq1, Seq2)Seq1 и Seq2в первой и третьей строках и символы, представляющие оптимальное локальное выравнивание между ними во второй строке. Символ | указывает аминокислоты или нуклеотиды, которые точно совпадают. Символ : обозначает аминокислоты или нуклеотиды, которые являются родственными, как определено матрицей оценки (несопоставленные с нулевым или положительным значением матрицы оценки).
[ возвращает вектор индексов 2 на 1, указывающий начальную точку в каждой последовательности для выравнивания.Score, Alignment, Start] = swalign(Seq1, Seq2)
... = swalign( требования Seq1,Seq2, ...'PropertyName', PropertyValue, ...)swalign с необязательными свойствами, использующими пары имя/значение свойства. Можно указать одно или несколько свойств в любом порядке. Каждый PropertyName должен быть заключен в одинарные кавычки и не учитывать регистр. Эти пары имя/значение свойства следующие:
... = swalign( указывает тип последовательностей. Варианты: Seq1,Seq2, ...'Alphabet', AlphabetValue)'AA' (по умолчанию) или 'NT'.
... = swalign( определяет матрицу оценки, используемую для локальной трассы. Значение по умолчанию:Seq1,Seq2, ...'ScoringMatrix', ScoringMatrixValue, ...)
'BLOSUM50' - Когда AlphabetValue равняется 'AA'
'NUC44' - Когда AlphabetValue равняется 'NT'
... = swalign( задает масштабный коэффициент, применяемый к выходному баллу, тем самым управляя единицами выходного балла. Выбор - это любое положительное значение.Seq1,Seq2, ...'Scale', ScaleValue, ...)
... = swalign( определяет штраф за открытие промежутка на трассе. Выбор - это любое положительное значение. По умолчанию: Seq1,Seq2, ...'GapOpen', GapOpenValue, ...)8.
... = swalign( определяет штраф за расширение зазора с использованием аффинной схемы штрафов за разрыв. Выбор - это любое положительное значение. Seq1,Seq2, ...'ExtendGap', ExtendGapValue, ...)
... = swalign( управляет отображением пространства оценки и траектории выигрыша трассы. Варианты: Seq1,Seq2, ...'Showscore', ShowscoreValue, ...)true или false (по умолчанию).
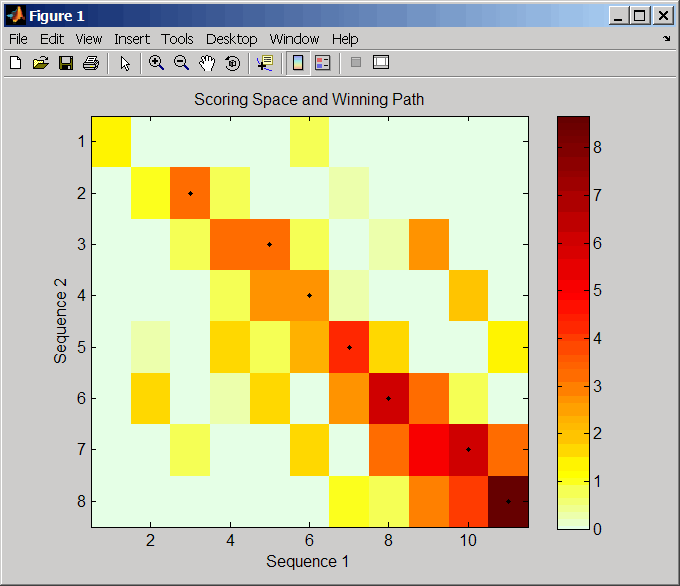
Пространство оценки представляет собой тепловую карту, отображающую наилучшие оценки для всех частичных выравниваний двух последовательностей. Цвет каждого (n1,n2) координата в пространстве оценки представляет наилучшую оценку для спаривания подпоследовательностей Seq1(s1:n1) и Seq2(s2:n2), где n1 является позицией в Seq1, n2 является позицией в Seq2, s1 является любой позицией в Seq1 между 1:n1, и s2 является любой позицией в Seq2 между 1:n2. Наилучший балл для спаривания конкретных подпоследовательностей определяется путем оценки всех возможных выравниваний подпоследовательностей путем суммирования совпадений и штрафов за разрыв.
Выигрышный путь представлен черными точками в скоринговом пространстве и иллюстрирует спаривание позиций в оптимальном локальном выравнивании. Цвет последней точки (справа внизу) победного пути представляет оптимальную оценку локального выравнивания для двух последовательностей и представляет собой Score вывод, возвращенный swalign.
Примечание
Пространство оценки визуально показывает тандемные повторы, небольшие сегменты, которые потенциально выравниваются, и частичные выравнивания доменов из переупорядоченных последовательностей.
Локально выровнять две аминокислотные последовательности, используя BLOSUM50 (по умолчанию) матрица оценки и значения по умолчанию для GapOpen и ExtendGap свойства. Возвращает оптимальную оценку локального выравнивания в битах и массив символов выравнивания.
[Score, Alignment] = swalign('VSPAGMASGYD','IPGKASYD') Score = 8.6667 Alignment = PAGMASGYD | | || || P-GKAS-YD
Локально выровнять две аминокислотные последовательности, определяющие PAM250 матрица оценки и открытый штраф за разрыв 5.
[Score, Alignment] = swalign('HEAGAWGHEE','PAWHEAE',... 'ScoringMatrix', 'pam250',... 'GapOpen',5) Score = 8 Alignment = GAWGHE :|| || PAW-HE
Локально выровнять две аминокислотные последовательности, возвращающие Score в единицах nat (nats) путем задания масштабного коэффициента log(2).
[Score, Alignment] = swalign('HEAGAWGHEE','PAWHEAE','Scale',log(2)) Score = 6.4694 Alignment = AWGHE || || AW-HE
[1] Дурбин, Р., Эдди, С., Крог, А. и Митчисон, Г. (1998). Анализ биологических последовательностей (Cambridge University Press).
Смит, Т. и Уотерман, М. (1981). Идентификация общих молекулярных подпоследовательностей. Журнал молекулярной биологии 147, 195-197.
aa2int | aminolookup | baselookup | blosum | dayhoff | gonnet | int2aa | int2nt | localalign | multialign | nt2aa | nt2int | nuc44 | nwalign | pam | pdbsuperpose | seqdotplot
