Визуализируйте и в интерактивном режиме исследуйте биологические последовательности
seqviewerseqviewer(Seq)seqviewer(Seq,Name,Value)seqviewer('close')seqviewer открывает приложение Sequence Viewer.
seqviewer( загружает последовательность Seq)Seq в приложение, где можно просмотреть и в интерактивном режиме исследовать последовательность.
seqviewer( открывает приложение с дополнительными опциями, заданными одним или несколькими аргументами пары Seq,Name,Value)Name,Value.
seqviewer( закрывает приложение Sequence Viewer. 'close')
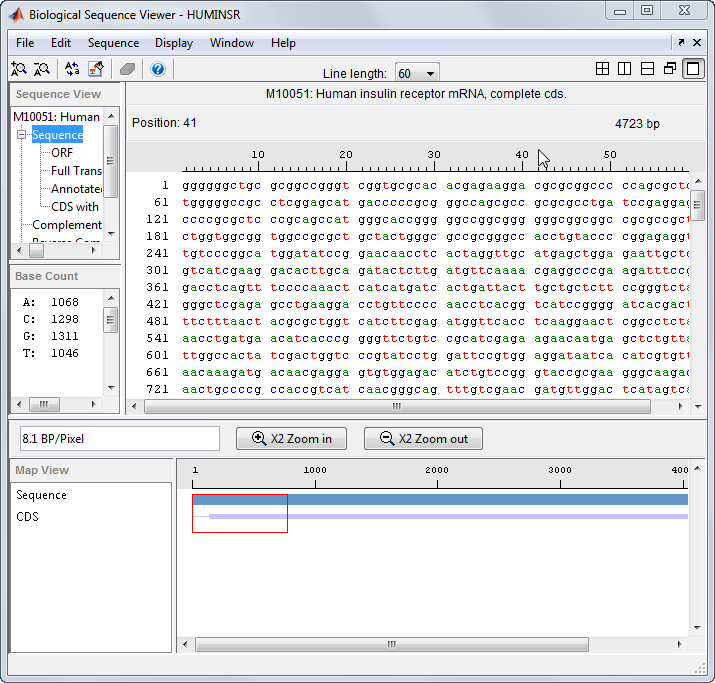
Получите последовательность из базы данных GenBank®.
S = getgenbank('M10051');Загрузите последовательность в приложение Sequence Viewer.
seqviewer(S)
Также можно нажать Sequence Viewer на вкладке Apps, чтобы открыть приложение и просмотреть биологическую последовательность S.
Закройте приложение.
seqviewer('close')aa2nt | aacount | aminolookup | basecount | baselookup | dimercount | emblread | fastaread | fastawrite | genbankread | geneticcode | genpeptread | getembl | getgenbank | getgenpept | nt2aa | proteinplot | seqcomplement | seqdisp | seqrcomplement | seqreverse | seqshoworfs | seqshowwords | seqwordcount
